
This tutorial is an introduction to working with sequence alignments and associated structures using Multalign Viewer. Data are taken from the enolase superfamily:
P.C. Babbitt, M.S. Hasson, J.E. Wedekind, D.R. Palmer, W.C. Barrett, G.H. Reed, I. Rayment, D. Ringe, G.L. Kenyon, and J.A. Gerlt, "The Enolase Superfamily: A General Strategy for Enzyme-Catalyzed Abstraction of the Alpha-Protons of Carboxylic Acids" Biochemistry 35:16489 (1996).
To follow along, first download the sequence alignment file super8.msf to a convenient location. This sequence alignment contains the barrel domains of eight enolase superfamily members.
On Windows/Mac, click the chimera icon; on UNIX, start Chimera from the system prompt:
unix: chimera
A basic Chimera window should appear after a few seconds. Choose File... Open, then locate and open super8.msf. Opening a sequence alignment file automatically starts Multalign Viewer and uses it to display the alignment. Multalign Viewer has its own set of preferences; from the Multalign Viewer (alignment window) menu, choose Preferences... Layout and change the font size and the sequence-wrapping behavior as desired. Size and place the sequence window and main Chimera window so that both are visible. If the sequence window becomes obscured at any point, it can be raised from the Chimera Tools menu (Tools... MAV - super8.msf... Raise).
| Multalign Viewer sequence window |
|---|
|
When the mouse focus is in the sequence window, the Page Down key (or space) moves the alignment view down and Page Up (or Shift-space) moves the alignment view up.
The names of the sequences are shown on the left; a Consensus sequence and Conservation histogram are shown above the multiple alignment. The superfamily is very diverse; there are very few positions in the alignment in which all the sequences have the same residue (indicated with a red capital letter in the consensus sequence).
The first sequence in the alignment, named mr, corresponds to the barrel domain of mandelate racemase from Pseudomonas putida. The next-to-the last sequence in the alignment, named enolyeast, corresponds to the barrel domain of enolase from Saccharomyces cerevisiae. There are multiple structures in the Protein Data Bank for each of these sequences; 2mnr (mandelate racemase) and 4enl (enolase) are used in this tutorial.
If you have internet connectivity, structures can be obtained directly from the Protein Data Bank. Choose File... Fetch by ID from the Chimera menu. In the resulting dialog, check the PDB option (if it is not already checked) and the option to Keep dialog up after Fetch. Fetch 2mnr, then 4enl, and then Close the dialog. If you do not have internet connectivity, instead download the files 2mnr.pdb and 4enl.pdb included with this tutorial and open them in that order with File... Open.
The view is initially centered on a protein colored white, the mandelate racemase structure. Off to the side and possibly out of view is the enolase structure, which is colored magenta. Adjust the view to contain both structures, and thicken the lines:
Actions... FocusThe structures are not matched in any way; the coordinates are taken straight from the Protein Data Bank. Move and scale the structures as desired throughout the tutorial. If you like, open the Side View (Tools...Viewing Controls... Side View).
Actions... Atoms/Bonds... wire width... 2
| sequence window after structure association |
|---|
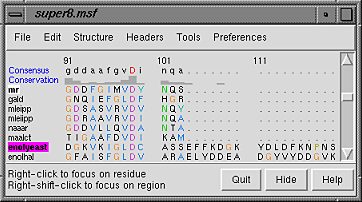 |
Notice that in the sequence window, the sequence names mr and enolyeast are now shown in bold within boxes colored white and magenta, respectively. This means that the sequences have been compared with the sequences of the structures, found to match sufficiently well, and automatically associated with the structures. The colors indicate which sequence has been associated with which structure.
The sequence alignment includes just the barrel domains; one might want to show only those parts of the structures. With the mouse, drag a box to highlight the entire sequence alignment. That will select the corresponding parts of the proteins. Show only those parts, and simplify the display to a chain trace:
Actions... Atoms/Bonds... show onlyBoxes drawn on the alignment are called regions. The active region is shown with a dashed outline. Delete the active region by clicking in some blank area of the sequence window (not on the sequences) to make sure this window has the mouse focus, then pressing the keyboard delete key. If the region is not active (the border is not dashed), first click on the region to activate it, then press delete.
Actions... Atoms/Bonds... backbone only... chain trace
Select... Clear Selection
Placing the cursor over any structure-associated residue in the sequence alignment shows the corresponding structure residue number near the bottom of the sequence window. For example, placing the cursor over the first residue in the mr sequence shows that it is associated with valine 134 in chain A of model #0 (the first structure opened).
The sequence alignment can be used to guide a structural match. From the Multalign Viewer menu, choose Structure... Match and click Apply without checking any boxes. This superimposes the structures using the alpha-carbons of all pairs of residues aligned in the sequence alignment. Readjust the view to focus on the structures:
Actions... FocusMatch statistics are reported briefly in the status line and are written to the Reply Log (Tools... Utilities... Reply Log). The match is fairly rough, giving an RMSD of 8.4 Å. Apparently, not all 175 pairs of residues in the same columns of the sequence alignment superimpose well in space. Try the structure matching again, but this time, turn on the option to Iterate by pruning... and edit the angstrom value to 1.0 before clicking OK. This improves the superposition of the most similar parts of the structures by omitting other parts from the fit.
Next, see where some of the conserved residues are within the structures. In the sequence window, find the first completely conserved residue in the alignment (the aspartate, D, at alignment position 99). Drag with the mouse to create a new region containing just that column of the alignment. The corresponding structure residues will be selected and can be displayed:
Actions... Atoms/Bonds... show
| active sites |
|---|
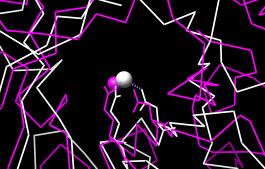 |
Create another region (this time, press Ctrl along with the mouse button to start a new region instead of replacing the first) for the next completely conserved residue. This is a glutamate, E, at alignment position 148; display it too:
Actions... Atoms/Bonds... showThe displayed residues point into the center of the barrel, ligating a catalytically important metal ion. Display the active site metal ions:
Select... Structure... ionsAlignment regions can also be created automatically. From the Multalign Viewer menu, choose Structure... Secondary Structure... show actual. This creates regions in the structure-associated sequences named structure helices (light yellow with gold outline) and structure strands (light green with green outline). A single region may consist of multiple disconnected boxes. The region names are listed in the Region Browser (Tools... Region Browser in the Multalign Viewer menu). Clicking on a region in the sequence window or clicking the Active checkbox for that region in the Region Browser will select the corresponding residues in any associated structures. For sequences not associated with structures, Structure... Secondary Structure... show predicted creates regions named predicted helices and predicted strands shown with gold and green outlines, respectively. Multiple regions can be deleted at once by choosing multiple lines in the Region Browser and clicking its Delete button. Close the Region Browser if it is open.
Actions... Atoms/Bonds... show
Select... Clear Selection
| conservation shown with color |
|---|
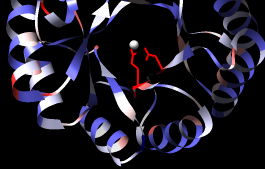 |
A structure can be colored according to the conservation in an associated sequence alignment. First, choose Tools... General Controls... Command Line from the main Chimera menu and use commands to close one of the structures and clear any selections:
Command: close 1Show a ribbon for the remaining structure:
Command: ~select
Command: ribbon @/displayThe @/display argument specifies only the part with currently displayed atoms, i.e., the barrel domain instead of the whole structure.
Choose Structure... Render by Conservation from the Multalign Viewer menu. The resulting Render by Attribute tool shows a histogram of the residue attribute mavPercentConserved, the percent conservation of the most prevalent residue at the corresponding position in the alignment. Adjust the coloring sliders on the histogram (and their Color values, if desired) before clicking Apply. Coloring the structure by mavPercentConserved shows the high conservation of the metal-binding residues and the low conservation of most residues around the outside of the barrel.
From the Attribute pulldown menu in the Render by Attribute dialog, choose the residue attribute named mavConservation. The values of this attribute are also shown in the sequence window Conservation line. Several different methods for calculating conservation are available. The Multalign Viewer Analysis preferences (Preferences... Analysis) control which method is used. If you wish, change the Conservation style to AL2CO and see how the Conservation histogram in the sequence window changes as any of the AL2CO parameters are varied. In Render by Attribute, it is necessary to update the values with Refresh... Values before recoloring the structure.
Multalign Viewer includes many features, only a few of which are sampled here. Users are encouraged to explore the menus and documentation for more information.
End the Chimera session:
Command: stop really