New Chimera Fitting Capabilities
Tom Goddard
June 27, 2011
Demonstration of new tools in UCSF Chimera software for fitting atomic models in
electron microscopy maps.
Note: Demo needs internet connection to use web services:
Fetch PDB/EMDB, BLAST, Modeller and SAXS.
What can go wrong in fitting?
- Map is wrong, for example, blending multiple conformations.
- Individual molecules from x-ray/nmr have the wrong conformation.
- Fail to find the best packing of the molecules in the map, especially at low resolutions.
Will show three new fitting tools that help with packing molecules in low
resolution maps.
Will show ways to examine the range of conformations available
from x-ray and nmr models.
Comparing fits of a map to small-angle x-ray scattering (SAXS) profiles can
indicate if the map shape is wrong.

Demonstration system: Alpha Crystallin
Alpha crystallin is a molecular chaperone that prevents protein
aggregation. There are two forms A and B with 60% sequence identity
with the A form found only in the vertebrate eye lens. It is about 40
percent of the protein in the human eye lens and prevents
crystallization of the other proteins in the lens. The EM map we will
look at is of the B form found it brain, heart and muscle tissue and
has a role in diseases such as Alzheimer's, Parkinson's and
multiple sclerosis where protein aggregation occurs.
Alpha crystallin forms oligomers of variable size, the map we will look
at consisting of 24 copies.
Demonstration Outline
- BLAST PDB.
- Multiple sequence and structure alignment.
- Creating a homology model.
- Molecular interface in crystal unit cell.
- Global search fitting.
- Determining map symmetry.
- Sequential molecule fitting to avoid clashes.
- Fitting with symmetry.
- Comparing models with small-angle x-ray scattering profiles.
Finding alpha crystallin PDB models
Use white background, silhouette edges and glossy lighting.
- Open human alphaB crystallin sequence (cryAB_seq.fasta, UniProt P02511).
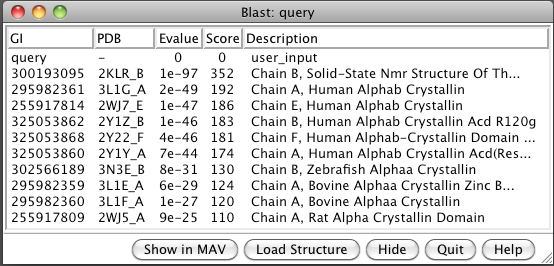
- BLAST sequence against PDB (Sequence dialog / Tools / Blast Protein).
Note 6 human, one zebrafish, two bovine, one rat.
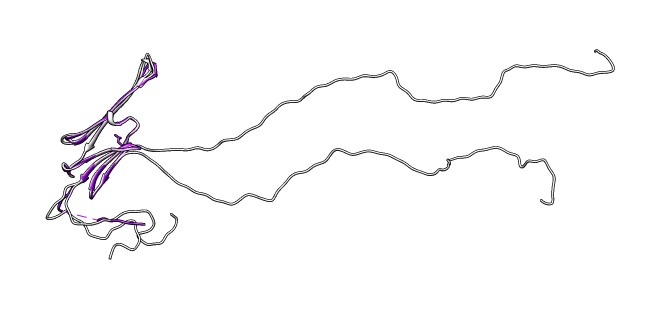
- Select 3L1G from list and load structure. Two beta sheets.
- Show multiple sequence alignment (Blast dialog, Show in MAV button).
- Load all 10 structures (Blast dialog, Load Structure button).
Homology modeling
- Create a homology model (MultAlign Viewer / Structure / Modeller Tools).
- Choose template 3L1F (bovine), 60% sequence identity.
- Under advanced options choose 2 models to output instead of 5 to speed it up.
- Note Modeller is run on web server unless you download it, and you need a free license key.
- Close all models except 3L1F and modeller results.
- Note long N-terminal region is not part of crystal structure.
- Delete N-terminal, command "del :1-61".
Binding Interfaces - Crystal Unit Cell
- Show binding interfaces from crystal packing.
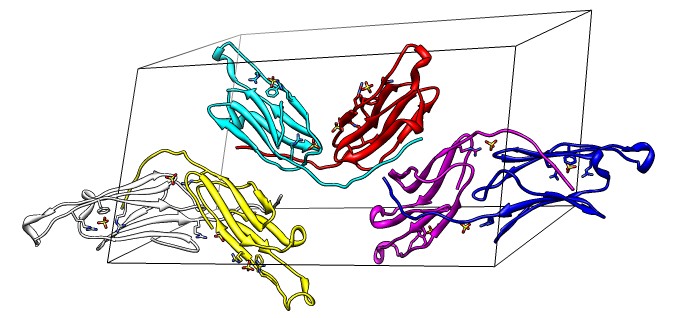
- Close session. Open human alphaB crystallin 3L1G.
- Show unit cell (Tools / Higher-Order Structure / Unit Cell).
- Show outline box.
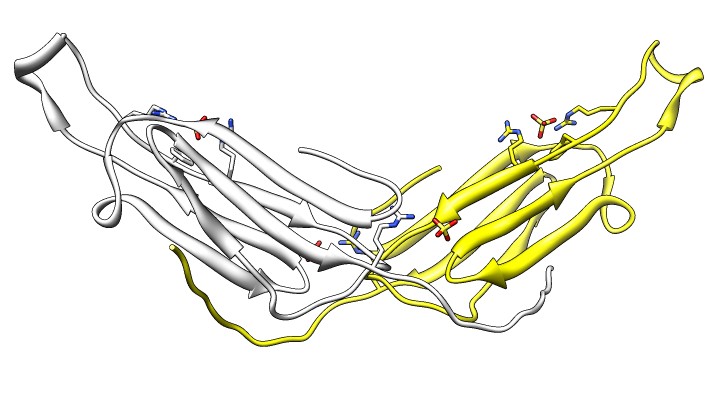
- Note 3 equivalent dimers. C-terminal lies in a groove at the edge of
the two beta sheets. Dimer is bound by each C-terminal tail sitting in
the groove of the other monomer.
- Another dimerization mode crosses 2 unit cells. Show it.
(Unit Cell / Options / Number of Cells "2 1 1")
- Each monomer has 3 binding sites: C-terminal tail, beta sheet groove,
and beta-sheet edge-to-edge extension (opposite edge from groove).
- Having 3 binding sites allows oligomers to form cages.
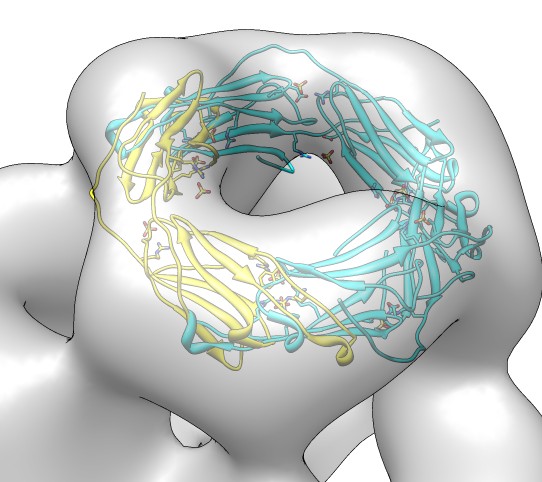
Fitting in EM map, global search
- Fit dimer instead of monomer because the map has 12-fold symmetry
but contains 24 copies of the monomer.
- Joined beta-sheet dimer would be better than C-terminal groove dimer
because it likely has less flexibility.
- We'll use the C-terminal dimer to better illustrate fitting with clashes
between monomers.
- Close all 3L1G monomers except white/yellow dimer using Model Panel.
- Easier to fit dimer as a single molecule. Command "combine #0,3".
Close original models.
- Rename combination to "3L1G dimer" using Model Panel / Rename button.
- Fetch alpha crystallin map, EMDB 1776. Negative stain, 20 Angstrom
resolution, tetrahedral symmetry.
- Fit search 10 placements, command "fit #1 #0 search 10".
- Show map as transparent. Also use one transparent layer.
Color model yellow with color button in model panel.
- Fit tried 10 random placements and orientations and did local
rigid body optimization.
- Many equivalent fits are listed due to symmetry.
Assigning and Using Map Symmetry
- Map symmetry is unfortunately not kept in map header. Use command
"measure symmetry #0" to deduce symmetry.
- Note status line says tetrahedral with center grid point 75 75 74.
- Recompute fits. Add "res 20" to get correlation options.
"fit #1 #0 search 10 res 20".
- Note only 4 unique positions found, identical fits removed. Hits column
of table says how often a fit was found. All fits found are moved to a single asymmetric unit.
- Close original fits.
- Note we have fits and 180 degree flip about dimer symmetry axis.
- Show clash volume between symmetric copies (Fit List / Options).
- 0.6 clash means 60% of volume of fit monomer is overlapped by symmetrically
placed monomers. Is using large envelope of simulated map. Show sim map.
- Show clashing monomers, "sym #1 group #0 update true".
Avoiding fitting clashes: sequential fitting
- Show only 3 dimers of one 3-fold donut.
Command "sym #1 group #0 range 50".
- Hide map to better see clashes.
- Fit sequentially. Command "fit #1,2.1,2.2 #0 seq 3 res 20" fits each
in turn subtracting the other two from the density first.
- Repeat last command to get better convergence.
- Repeat again with "seq 15" for 5 rounds of fitting each monomer.
- This protocol was taken from an Alasdair Steven's lab paper on Rous sarcoma
virus.
Avoiding fitting clashes: symmetric fitting
- Above method is a poor approach for symmetry related monomers since
it does not guarantee symmetry. It is better applied to proteins that are
not related by symmetry.
- Better approach to this problem is to fit one monomer taking account
of clashes of symmetrically placed monomers.
- Fit full symmetric assembly. Restore best fit from fit list.
Remove symmetry copies, command "~sym". Fit command "fit #1 #0 res 20 sym true".
- Symmetric fitting optimizes the correlation of the full symmetric assembly by moving one monomer and keeping all other virtual monomers related by symmetry.
- This type of fitting has been done by the URO program for many years
although it does it in Fourier space while we do it in real space.
- Note reported correlation 0.95.
- This avoids clashes because if two monomers overlap they create double
density that gives poor correlation with experimental map. Clashes are
implicitly avoided -- there is no special repulsion introduced.
Small-angle x-ray scattering profiles
- Small-angle x-ray scattering (SAXS) data can be used to independently
verify a model obtained by fitting EM data.
- I don't have experimental SAXS data for the alphaB crystallin EM map,
so I'll show an example where I have data.
- SAXS data from Stephen Weeks in Sergei Strelkov's lab looks at
alphaB crystallin dimers.
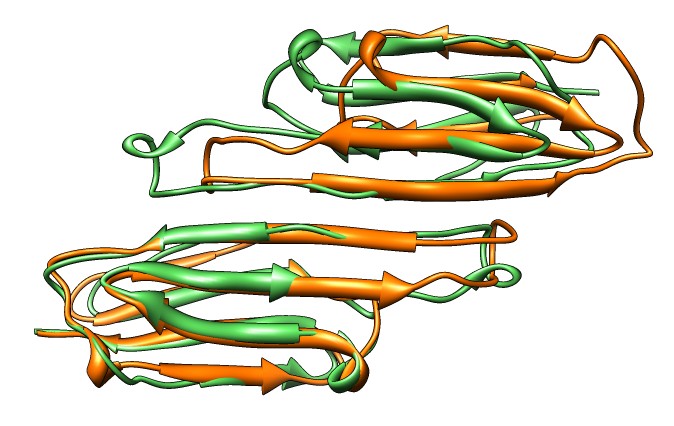
- Compare SAXS data to joined beta-sheet dimers showing different
strand registrations in different crystal structures,
3L1G (gray) and 2WJ7 (purple). Open session 3l1g_2wj7_dimer_registration.py.
- Use SAXS dialog (Tools / Higher-Order Structure) to compute profiles
for 3L1G and 2WJ7 fit to experimental data B1_reduced.dat.
- 2WJ7 registration fits better. Strelkov et. al. found only this
registration at physiological pH but other registrations at lower pH
used for some crystal structures.
Published SAXS dimer result, Strelkov lab
SAXS fit of alphaB crystallin dimers with different beta-sheet registrations
from
Baranova EV, Weeks SD, Beelen S, Bukach OV, Gusev NB, Strelkov SV.
Three-Dimensional Structure of α-Crystallin Domain Dimers of Human Small Heat Shock Proteins HSPB1 and HSPB6.
J Mol Biol. 2011 May 30.

Fig. 5 from Strelkov lab paper.
"Fitting of the obtained SAXS data with atomic models. (a) Scattering
curves of reduced B1α, oxidized B1α and B6α were fitted with the
coordinates of the ACD dimer of αB-crystallin (PDB ID: 2WJ7) using
CRYSOL.53 For clarity, the curves are offset in vertical
direction. Experimental and calculated scattering curves are shown in
black and red, respectively. The calculated quality of fit, χ2, is
given. (b) Scattering curve of the reduced B1α sample was fitted with
various dimeric ACD structures: bovine αB-crystallin (API), human
αB-crystallin (APII), rat HSPB6 (APIII) and human αB-crystallin
determined by NMR (NMR APII). (c) Ribbon diagrams of the atomic models
used for fitting in (b). The orientation of the right-hand ACD is the
same for all models."
Acknowledgements

- Eric Pettersen - Sequence and structure analysis.
- Elaine Meng - Email help, documentation and tutorials.
- YZ Yang Zheng - Homology modeling, SAXS.
- Greg Couch - OpenGL graphics.
- Darren Weber - Animation.
- Conrad Huang - Web services and project leader.
- Tom Ferrin - Head of lab.
|
- Dina Schneidman - SAXS
- Keren Lasker - MultiFit
- Ben Webb - Modeller
- Andrej Sali
- Stephen Weeks - SAXS data
- Sergei Strelkov
|