
MinRMS is a stand-alone, nongraphical program that generates structural alignments (superpositions) of the proteins in two input PDB files. Minrms Plot is a Collaboratory-aware extension of Chimera used to display and analyze the results. The alternative, AlignPlot, has additional features, but is not Collaboratory-aware.
Briefly, MinRMS creates many superpositions of the same two structures differing in the number of residues paired; for a given number of pairings, the superposition is that which minimizes the RMSD. The nth entry of the "lowest-RMSD list" consists of a structural alignment and the n residue pairings within it used to calculate the RMSD. Each list item can be expressed as a sequence alignment in MSF format (file alignn.msf); the corresponding rotation/translation information is included in the comments. The n residue pairings are aligned in sequence within the MSF file. Multalign Viewer or MSF Viewer can be used to display the sequence alignments while the structural alignments are being viewed with Minrms Plot (or AlignPlot).
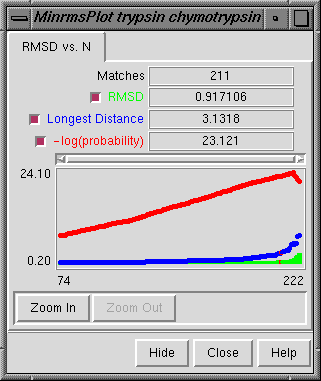
There are several ways to start Minrms Plot, a tool in the Homology category. Starting Minrms Plot brings up a dialog box requesting the file align_chimera.info previously created by MinRMS. This file lists pdb1 and pdb2 (the names of the two PDB files), the name of the plot data file (currently just align_chimera.plot), and the number of alignment files to be read.
Upon reading the file, Chimera will display the molecules in pdb1 and pdb2 in the graphics window and open an additional MinrmsPlot window containing a graph. Initially, the molecules are not superimposed (and may not both be visible); their positions depend on the coordinates in the input PDB files. Showing just the backbone or chain trace of proteins is recommended to simplify the display.
|
Zoom In expands the graph to show a smaller range of N in the same area; Zoom Out reverses this. Close closes the MinrmsPlot window, and Help opens this manual page in a browser window.
The structure in pdb1 is static (unless moved interactively by the user) and the structure in pdb2 is reoriented relative to the first. Residue pairings are indicated with green pseudobonds in the graphics window:

|